Pr Mathieu Molimard- Département de pharmacologie- CHU de Bordeaux
Céline Laffont Fév 2011
Définitions
Pharmacodynamie (PD) → Action du médicament sur l’organisme
Pharmacocinétique (PK) → Action de l’organisme sur le médicament
La pharmacocinétique (PK) est l’étude de l’exposition de l’organisme au médicament en vue d’étudier ses relations avec les
effets du médicament (PK/PD).
La PK est l’étude de la cinétique de l’absorption, de la distribution, du métabolisme et de
l’excrétion des médicaments.
Paramètres pharmacodynamiques
| Notions | Paramètres PD |
|---|---|
| 1- Efficacité |
1- Intensité
de l ’effet : effet maximum |
| 2- Puissance |
2- Concentration
capable de produire un effet donné : 50% de l’effet maximum |
| 3- Sélectivité/sécurité |
3- Index thérapeutique : rapports entre des conc. thérapeutiques ou toxiques |
Paramètres pharmacocinétiques
| Etapes | Paramètres PK |
|---|---|
| 1- Absorption | 1- Biodisponibilité |
| 2- Distribution | 2- Volume de distribution, % de liaison aux protéines plasmatiques |
| 3- Métabolisme | 3 &4- Clairance |
| 4- Excrétion/Elimination | 4- Temps de demi-vie |
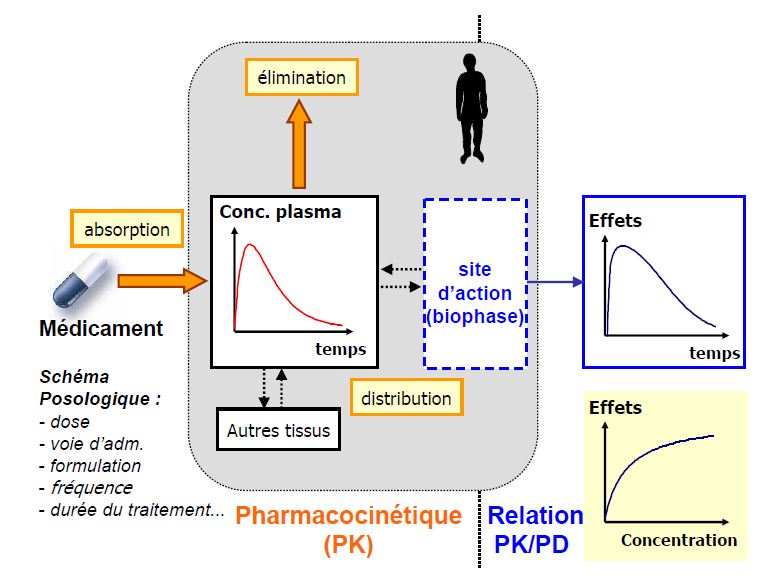
La relation qui lie pharmacocinétique et pharmacodynamie
Pharmacocinétique de la Voie IV
Absorption- Distribution
- Métabolisation
- Elimination
A.D.M.E.

Source de variabilité inter- et intra individuelle car influencées par de nombreux facteurs
ABSORPTION
Se réfère ici à l’absorption « systémique »
Etapes du devenir du médicament qui conduisent le produit administré de son site d’administration jusqu’à la circulation générale (sang)
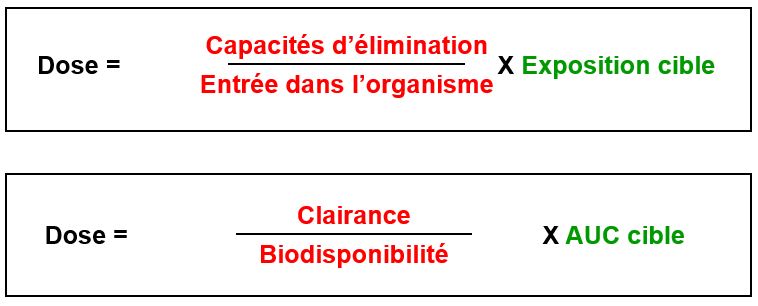
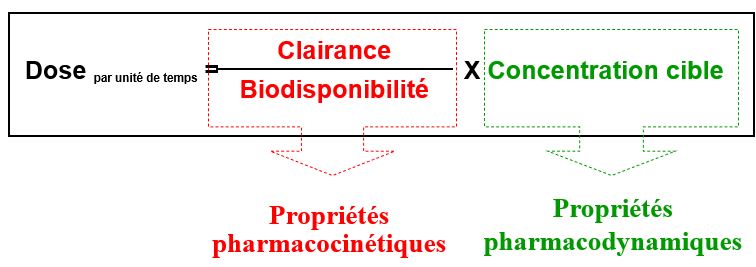
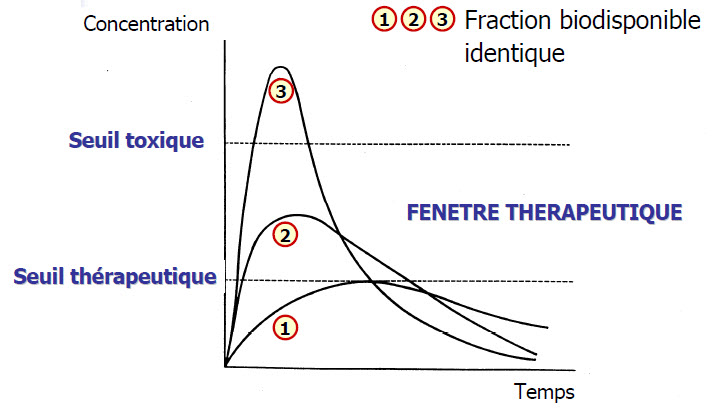
Biodisponibilité (F)
Fraction de la dose de médicament administré qui parvient à la circulation générale et la vitesse avec laquelle elle y parvient
Voie IV = Pas d’absorption
- Elimine un facteur de variabilité (de sécurité)
- Biodisponibilité 100%
- Dose différente de per os
- Pas de premier passage hépatique
Notion de vitesse
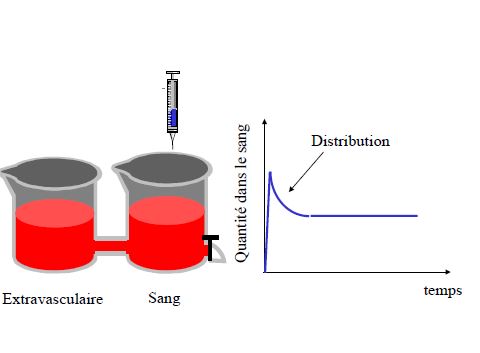
DISTRIBUTION
Répartition du produit administré dans l’organisme à partir de la circulation générale
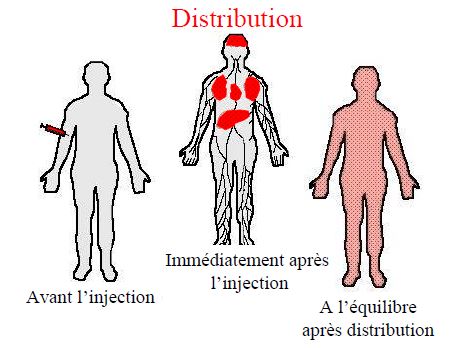
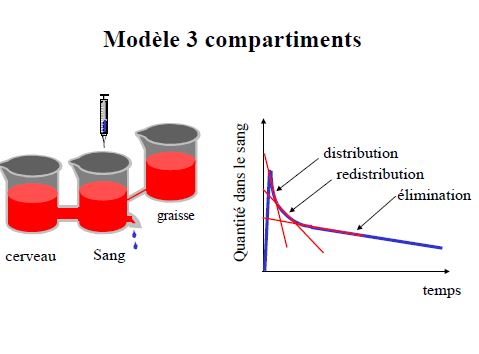
Modèles à 3 compartiments
- plasma
- organes très vascularisés (cœur foie rein cerveau)équilibre rapide
- organes peu vascularisés (graisse, tendons cartilage)équilibre lent
Volume de distribution Vd
Constante de proportionnalité entre la quantité de substance présente dans l’organisme et la concentration plasmatique

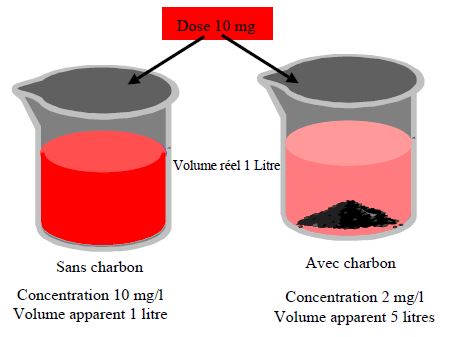
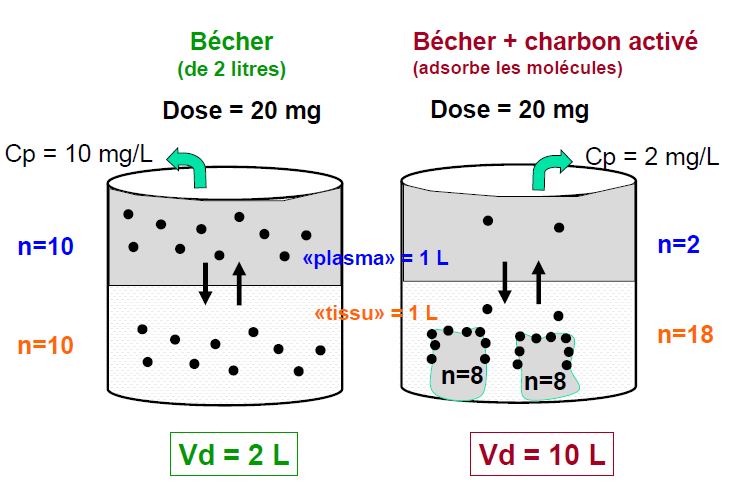
Vd grand concentration tissulaires élevée/plasma distribution non homogène
Vd petit Au minimum volume plasmatique =0,04 l/kg
Liaison aux protéines
Seule la concentration libre est active
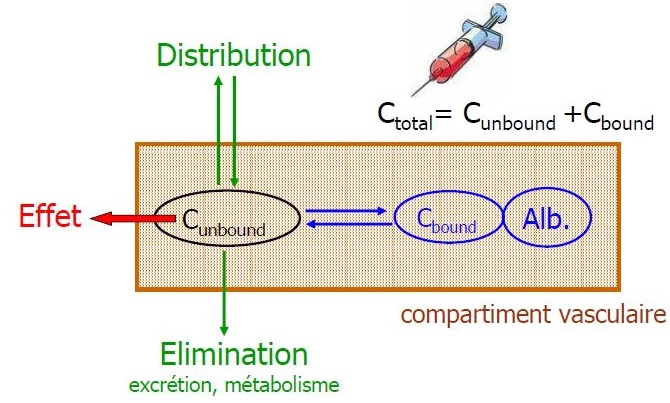
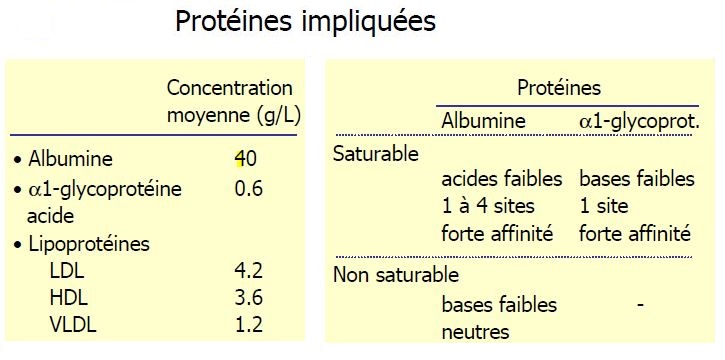
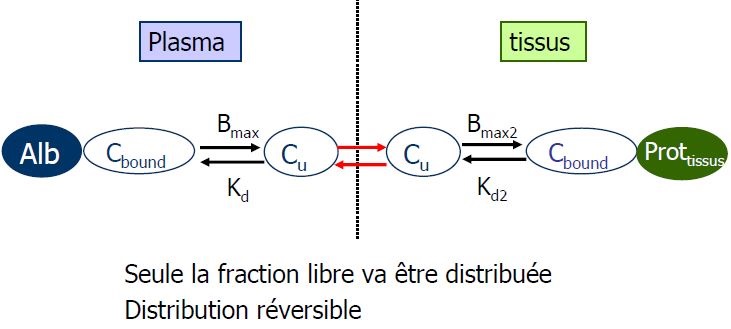
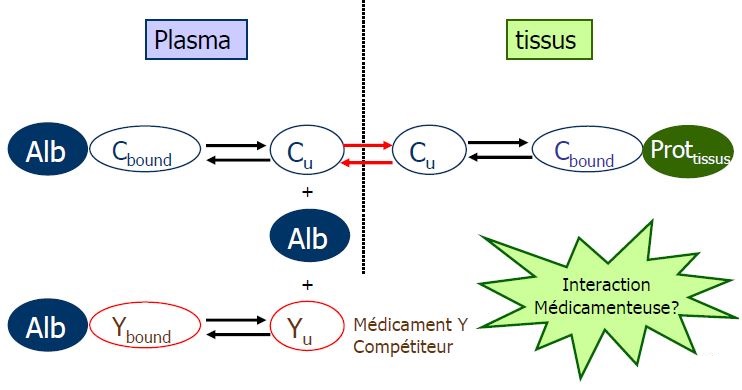
Variation de la fixation
- Acides faibles ont une fixation saturable d'où interaction
- NN interaction bilirubine
- Age : hypoalbuminémie
- IR, IHC : hypoalbuminémie + modification structure albumine
+ compétition avec métabolites
Flux sanguin tissulaire adulte
Débit cardiaque 5400 ml/min
Foie 1700 ml/min
Rein 1000 ml/min
SNC 800 ml/min
Graisse 250 ml/min
MÉTABOLISATION
Phases
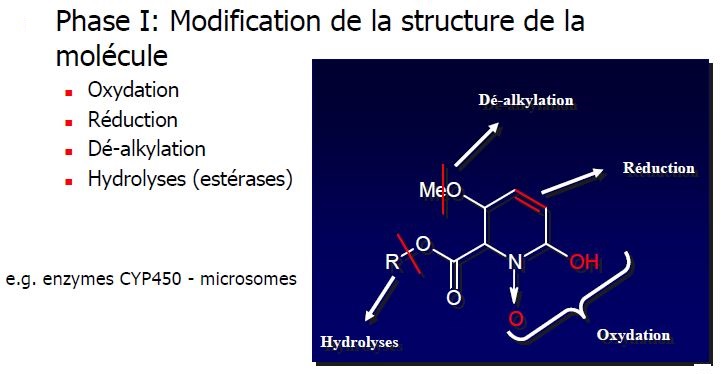
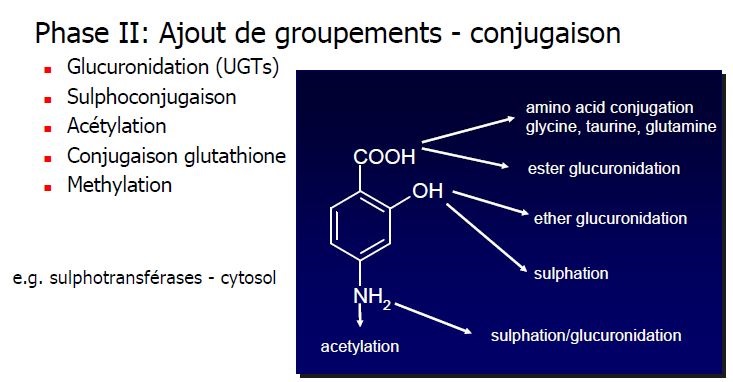
Lieux
Foie principalement
autres tissus
Variations
polymorphismes, IHC, compétition
Interractions médicamenteuses
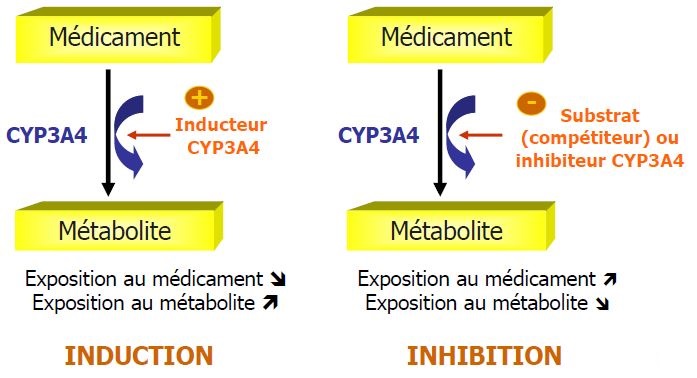
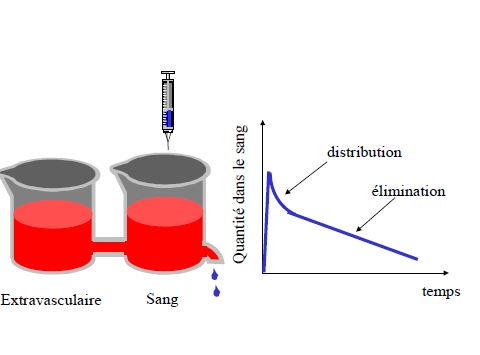
ÉLIMINATION
Clairance = volume de plasma épuré de la substance par unité de temps
Clairance totale = somme des clairances = dose/AUC
Clairance rénale = fraction de clairance totale = fe x clairance totale
(fe=quantité cumulative dans les urines/quantité administrée)
Clairance hépatique = clairance biliaire + clairance métabolique
Clairance
Elimination d’ordre 1 =Clairance constante (le + fréquent dans les doses thérapeutiques)
L’élimination est proportionnelle à la quantité présente dans l’organisme:
Débit d’élimination = Cl x C
Elimination non linéaire = Clairance variable
Facteurs de variation de la clairance
Elimination à capacité limitée
par activité enzymatique saturable : alcool (au dela de 0,5g/l)
phénytoïne
toxicocinétique
Facteurs de variation de la clairance
- Fonction rénale ou hépatique
- Flux sanguin dans l’organe
- Fraction libre
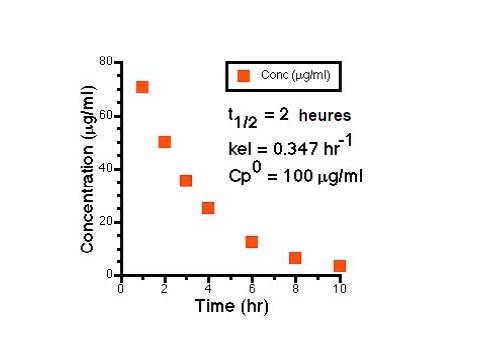
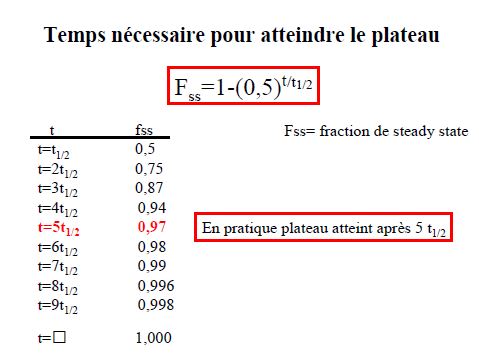
Demi-vie d’élimination
T1/2 temps nécessaire pour que la concentration plasmatique diminue de moitié
La demi-vie dépend de la clairance et du volume de distribution
T1/2= 0,693xVd/Cl
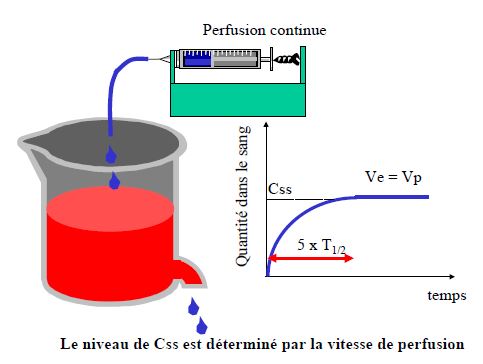
Le produit peut être considéré comme virtuellement éliminé après 5 demi-vies
t fraction éliminée
t=t1/2 0,5
t=2t1/2 0,75
t=3t1/2 0,87
t=4t1/2 0,94
t=5t1/2 0,97
t=6t1/2 0,98
t=7t1/2 0,99
t=8t1/2 0,996
t=9t1/2 0,998
t= 1,000
Intérêt de la T1/2
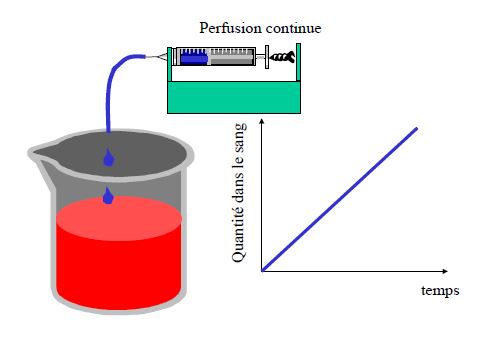
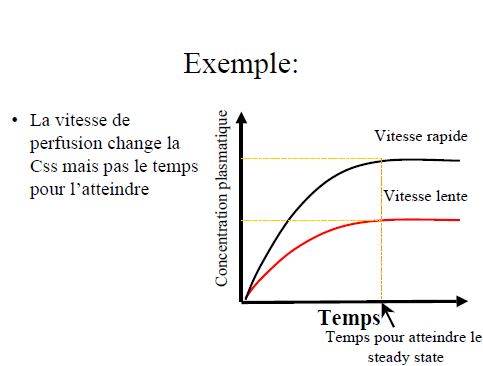
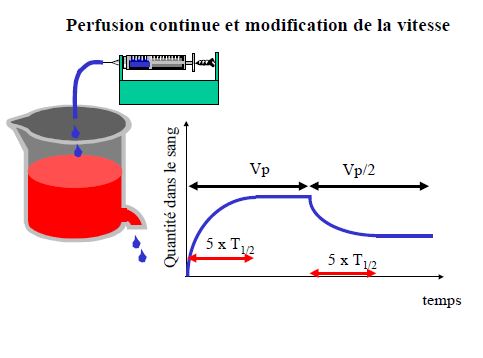
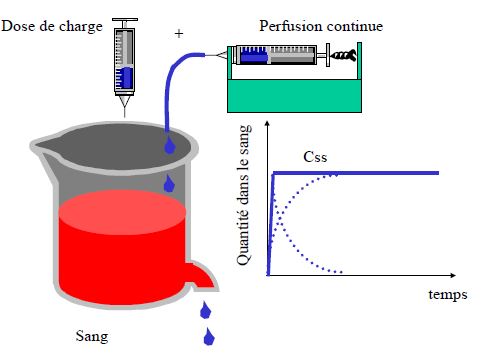
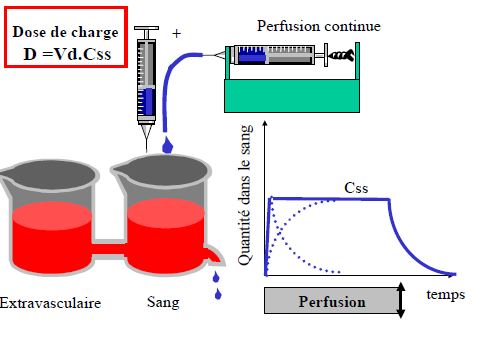
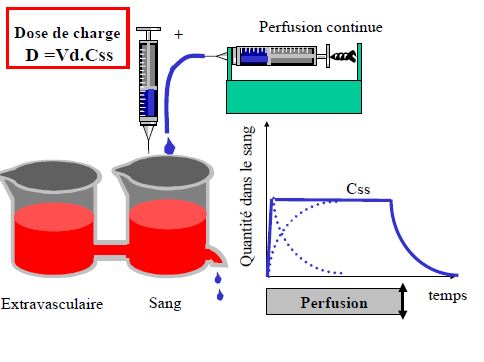
- Prédire le temps nécessaire pour atteindre l’état d’équilibre (steady-state) lors d’une perfusion ou de prises répétées
- Prédire l’accumulation après prises répétées
Paramètres PK
| Etapes |
Paramètres PK |
|---|---|
| 1 - Absorption | 1 - Biodisponibilité |
| 2 - Distribution | 2 - Volume de distribution, % de liaison aux protéines plasmatiques |
| 3 - Métabolisme | 3,4 - Clairance |
| 4 - Excrétion/Elimination | 2,4 – Temps de demi-vie |
Pharmacocinétique en administration unique
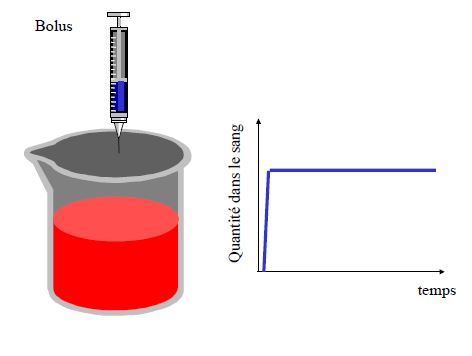
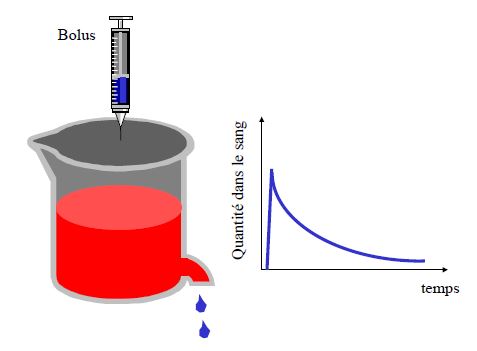
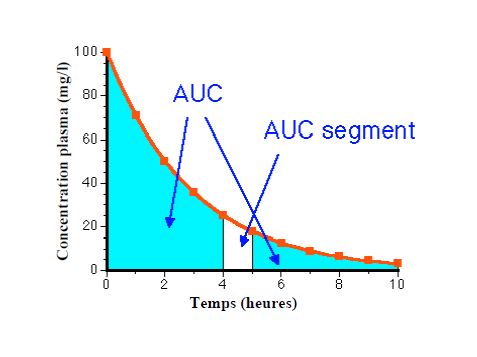
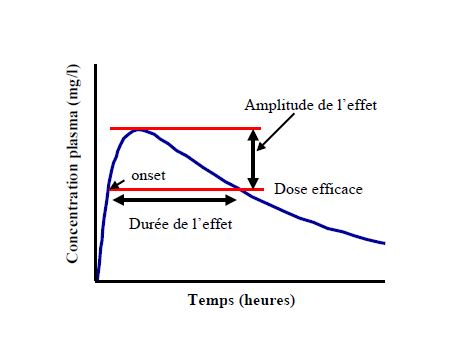
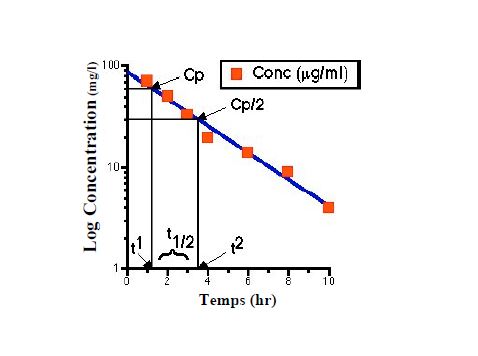
La substance peut être considérée comme éliminée après 5 demi-vies
Pharmacocinétique en administration répétée
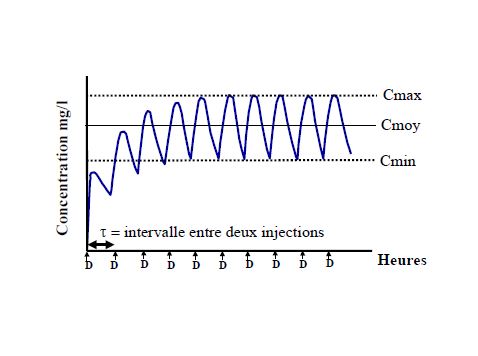

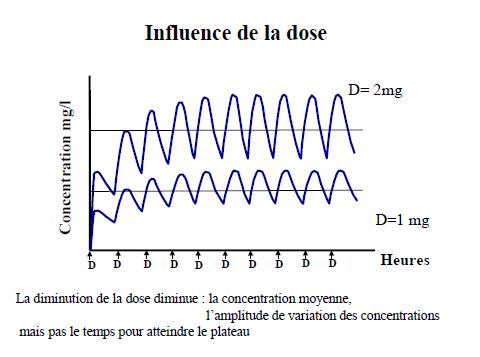
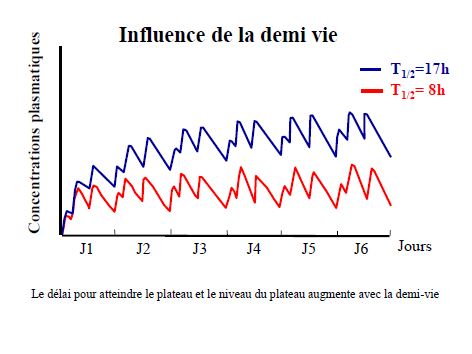
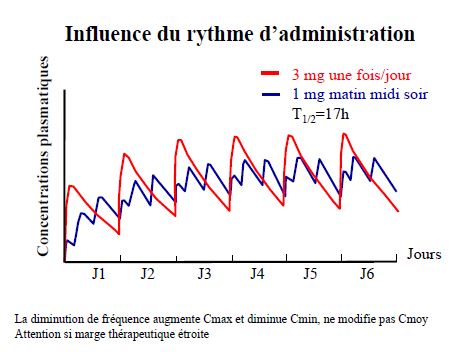
Pharmacocinétique en administration continue